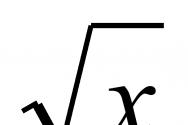Просто о сложном: что такое масс-спектрометрия, или как взвесить молекулу. Масс-спектрометрия: ее виды и особенности Достоинства масс спектрометрии
- Введение
- Краткая история масс-спектрометрии
- Ионизация
- Масс-анализаторы
- Детектор
- Природная и искусственная изотопия
- Масс-спектрометры для изотопного анализа
- Скорость сканирования
- Разрешение
- Динамический диапазон
- Чувствительность
- Какие бывают масс-спектрометры
Итак, масс-спектрометры используются для анализа органических соединений и неорганических.
Органические вещества в большинстве случаев представляют собой многокомпонентные смеси индивидуальных компонентов. Например, показано, что запах жареной курицы составляют 400 компонентов (то есть, 400 индивидуальных органических соединений). Задача аналитики состоит в том, чтобы определить сколько компонентов составляют органическое вещество, узнать какие это компоненты (идентифицировать их) и узнать сколько каждого соединения содержится в смеси. Для этого идеальным является сочетание хроматографии с масс-спектрометрией. Газовая хроматография как нельзя лучше подходит для сочетания с ионным источником масс-спектрометра с ионизацией электронным ударом или химической ионизацией, поскольку в колонке хроматографа соединения уже находятся в газовой фазе. Приборы, в которых масс-спектрометрический детектор скомбинирован с газовым хроматографом, называются хромато-масс-спектрометрами.
Многие органические соединения невозможно разделить на компоненты с помощью газовой хроматографии, но можно с помощью жидкостной хроматографии. Для сочетания жидкостной хроматографии с масс-спектрометрией сегодня используют источники ионизации в электроспрее (ESI) и химической ионизации при атмосферном давлении (APCI), а комбинацию жидкостных хроматографов с масс-спектрометрами называют ЖХ/МС или LC/MS по английски. Cамые мощные системы для органического анализа, востребованные современной протеомикой, строятся на основе сверхпроводящего магнита и работают по принципу ионно-циклотронного резонанса. Они также носят название FT/MS, поскольку в них используется Фурье преобразование сигнала.
Новый класс масс-спектрометров - это гибридные приборы. Гибридными их называют потому, что они, на самом деле, включают в себя два масс-спектрометра, по крайней мере один из которых, может работать как независимый прибор. Примерами таких приборов являются масс-спектрометр ионно-циклотронного резонанса FINNIGAN LTQ FT, в котором линейная квадрупольная ионная ловушка FINNIGAN LTQ может работать как индивидуальный прибор, детектирующий ионы после МС или МСn с помощью двух вторично-электронных умножителей, так и готовить и пересылать ионы в циклотронную ячейку, выталкивая их в направлении, параллельном оси квадруполя. Также гибридным является LTQ QRBITRAP, который работает совершенно аналогично. Преимущества таких схем очевидны, линейная ловушка обладает самой высокой чувствительностью, работает в режиме тандемной масс-спектрометрии с n до 10, осуществляет разнообразные интеллектуальные функции сканирований, а масс-спектрометр ионно-циклотронного резонанса и орбитальная ловушка ионов обладают высоким разрешением и могут с высочайшей точностью измерять отношения массы к заряду ионов. Для анализа элементного состава самыми привлекательными являются масс-спектрометры с индуктивно-связанной плазмой. С помощью этого прибора определяют из каких атомов составлено вещество. Этот же метод анализа может показывать и изотопный состав. Но лучше всего измерять изотопный состав с помощью специализированных изотопных приборов, регистрирующих ионы не на одном детекторе в разное время их прихода на него, а каждый ион на своем персональном коллекторе и одновременно (так называемое параллельное детектирование).
Однако, прежде чем перейти к приборам для измерения изотопного состава, кратко остановимся на том что такое изотопы.
Природная и искусственная изотопия Атомы состоят из ядра и электронных оболочек. Свойства атомов определяются тем, сколько протонов (положительно заряженных элементнарных частиц) содержит ядро. Ядро помимо протонов содержит и нейтроны. Природа распорядилась так, что при равном количестве протонов ядро может содержать разное количество нейтронов. Атомы с одинаковым количеством протонов в ядре, но с разным количеством нейтронов отличаются по массе на одну или несколько единиц атомной массы (а.е.м.) и называются изотопами. Большинство элементов имеют определенный набор стабильных изотопов. Радиоактивные изотопы не являются стабильными и распадаются с образованием стабильных изотопов. Природная распространенность изотопов для каждого элемента известна. Некоторые элементы в природе являются моноизотопными, то есть 100 % природной распространенности приходится на один изотоп (например, Al, Sc, Y, Rh, Nb и т.д.), а другие имеют множество стабильных изотопов (S, Ca, Ge, Ru, Pd, Cd, Sn, Xe, Nd, Sa и т.д.). В технологической деятельности люди научились изменять изотопный состав элементов с целью получения каких-либо специфических свойств материалов (например, U235 имеет способность к спонтанной цепной реакции и может использоваться в качестве топлива для атомных электростанций или атомной бомбы) или использования изотопных меток (например, в медицине).
Поскольку массы изотопов отличаются, а масс-спектрометрия измеряет массу, естественно, этот метод становится самым удобным для определения изотопного состава. В то же время, информация по изотопному составу помогает идентифицировать органические соединения и позволяет дать ответы на многие вопросы начиная от определения возраста пород для геологии и кончая определением фальсификатов многих продуктов и установлением места происхождения товаров и сырья.
Масс-спектрометры для изотопного анализа. Масс-спектрометры для определения изотопного состава должны быть очень точными. Для анализа изотопного состава легких элементов (углерод, водород, кислород. сера, азот и т.д.) используется ионизация электронным ударом. В этом случае годятся все методы ввода газовой фазы, как и в органических масс-спектрометрах (DELTA Plus ADVANTAGE, FINNIGAN DELTA Plus XL и FINNIGAN МАТ253).
Для анализа изотопов более тяжелых элементов используется термоионизация (FINNIGAN TRITON TI) или ионизация в индуктивно-связанной плазме c параллельным детектированием (FINNIGAN NEPTUNE, и одноколлекторным детектированием FINNIGAN ELEMENT2).
Практически во всех типах изотопных масс-спектрометров используются магнитные масс-анализаторы.
Характеристики масс-спектрометров и масс-спектрометрических детекторов
Важнейшими техническими характеристиками масс-спектрометров являются чувствительность, динамический диапазон, разрешение, скорость.
Скорость сканирования. Масс-анализатор, как мы показывали выше, пропускает ионы с определенным соотношением массы и заряда в определенное время (кроме многоколлекторных приборов и ионно-циклотронного резонанса, орбитальной ловушки ионов). Для того, чтобы проанализировать все ионы по отношению их массы к заряду он должен сканировать, то есть параметры его поля должны за заданный промежуток времени пройти все значения, нужные для пропускания к детектору всех интересующих ионов. Эта скорость разворачивания поля называется скоростью сканирования и должна быть как можно больше (соответственно, время сканирования должно быть как можно меньше), поскольку масс-спектрометр должен успеть измерить сигнал за короткое время, например за время выхода хроматографического пика, которое может составлять несколько секунд. При этом, чем больше масс-спектров за время выхода хроматографического пика будет измерено, тем точнее будет описан хроматографический пик, тем менее вероятно будет проскочить мимо его максимального значения, а с помощью математической обработки определить является ли он индивидуальным и «доразделить» его с помощью масс-спектрометрии.
Самым медленным масс-анализатором является магнит, минимальное время его сканирования без особой потери чувствительности составляет доли секунды (MAT 95XP). Квадрупольный масс-анализатор может разворачивать спектр за десятые доли секунды (TSQ QUANTUM), а ионная ловушка еще быстрее (POLARISQ, FINNIGAN LCQ ADVANTAGE MAX, FINNIGAN LCQ DECA XP MAX), линейная ионная ловушка - еще быстрее (LTQ) и чуть медленнее масс-спектрометр ионно-циклотронного резонанса FINNIGAN LTQ FT.
Инновационный квадрупольный хромато-масс-спектрометр FINNIGAN TRACE DSQ и его экономичный аналог FINNIGAN FOCUS DSQ способны сканировать со скоростью около 11,000 а.е.м. в секунду. Это открывает новые возможности, например, можно практически одновременно получать полный масс-спектр соединения для его однозначной идентификации и вести селективный мониторинг ионов (SIM), на несколько порядков понижающий предел обнаружения.
Любое сканирование всех перечисленных выше масс-анализаторов является компромиссным - чем больше скорость сканирования, тем меньше времени тратиться на запись сигнала на каждое массовое число, тем хуже чувствительность. Однако, для обычного анализа скорости квадрупольного анализатора или ионной ловушки достаточно. Другой вопрос, когда речь идет о высокопроизводительном анализе сложных матриц. В этом случае было бы хорошо воспользоваться сверхбыстрой хроматографией (на тонких коротких быстро прогреваемых колонках). Для такой задачи лучше всего подойдет времяпролетный масс-спектрометр (TEMPUS). Он способен записывать масс-спектры со скоростью 40,000 в секунду!
Разрешение. Наглядно разрешение (разрешающую способность) можно определить, как возможность анализатора разделять ионы с соседними массами. Очень важно иметь возможность точно определять массу ионов, это позволяет вычислить атомную композицию иона или идентифицировать пептид путем сравнения с базой данных, сократив число кандидатов с тысяч и сотен до единиц или одного единственного. Для магнитных масс-анализаторов, для которых расстояние между пиками масс-спектра не зависит от масс ионов, разрешение представляет собой величину равную M/DM. Эта величина, как правило, определяется по 10 % высоте пика. Так например, разрешение 1000 означает, что пики с массами 100.0 а.е.м. и 100.1 а.е.м. отделяются друг от друга, то есть не накладываются вплоть до 10 % высоты.
Для анализаторов, у которых расстояние между пиками меняется в рабочем диапазоне масс (чем больше масса, тем меньше расстояние), таких как квадрупольные анализаторы, ионные ловушки, времяпролетные анализаторы, строго говоря, разрешение имеет другой смысл. Разрешение, определяемое как M/DM в данном случае, характеризует конкретную массу. Имеет смысл характеризовать эти масс-анализаторы по ширине пиков, величине, остающейся постоянной во всем диапазоне масс. Эта ширина пиков, обычно, измеряется на 50 % их высоты. Для таких приборов ширина пика на полувысоте равная 1 является неплохим показателем и означает, что такой масс-анализатор способен различить номинальные массы, отличающиеся на атомную единицу массы практически во всем его рабочем диапазоне. Номинальной массой или массовым числом называют ближайшее к точной массе иона целое число в шкале атомных единиц массы. Например, масса иона водорода Н+ равна 1.00787 а.е.м., а его массовое число равно 1. А такие масс-анализаторы, которые, в основном, измеряют номинальные массы, называют анализаторами низкого разрешения. Мы написали «в основном», потому что сегодня есть и такие масс-анализаторы, которые формально относятся к низкоразрешающим, а на деле таковыми уже не являются. Высокая технология, прежде всего самого передового разработчика Thermo Electron, уже сегодня предложила на рынок аналитического оборудования высокоразрешающие квадрупольные приборы. Так например, новейший FINNIGAN TSQQuantum легко работает при ширине пика масс-спектра на полувысоте 0.1 а.е.м. Знающие люди могут возразить: «Но такую ширину пика можно получить на каждом квадрупольном масс-спектрометре!» И они будут правы, действительно, каждый квадруполь можно отстроить до этого уровня разрешения. Но что произойдет при этом с сигналом? При переходе от ширины пика на полувысоте в 1 а.е.м. к 0.1 а.е.м. величина сигнала на всех квадруполях упадет практически на два порядка по величине. Но не на TSQ Quantum , на нем она уменьшится всего в два с половиной раза. Ионные ловушки в узком диапазоне масс могут работать как масс-спектрометры высокого разрешения, обеспечивая, как минимум, разделение пиков, отстоящих на 1/4 а.е.м. друг от друга. Масс-спектрометры с двойной фокусировкой (магнитной и электростатической), ионно-циклотронного резонанса - приборы среднего или высокого разрешения. Типичным для магнитного прибора разрешением является >60,000, а работа на уровне разрешения 10,000 - 20,000 является рутинной. На масс-спектрометре ионно-циклотронного резонанса на массе около 500 а.е.м. можно легко достигнуть разрешения 500,000, что позволяет проводить измерения массы ионов с точностью до 4-5 знака после запятой. Разрешения в несколько тысяч также можно добиваться при использовании времяпролетных масс-анализаторов, однако, на высоких массах, в области которых, собственно этот прибор имеет преимущество перед другими, и этого разрешения хватает лишь для того, чтобы измерить массу иона с точностью +/- десятки а.е.м.Как видно из вышесказанного, разрешение тесно связано с другой важной характеристикой - точностью измерения массы. Проиллюстрировать значение этой характеристики можно на простом примере. Массы молекулярных ионов азота (N2+)и монооксида углерода (СО+) составляют 28.00615 а.е.м. и 27.99491 а.е.м., соответственно (оба характеризуются одним массовым числом 28). Эти ионы будут регистрироваться масс-спектрометром порознь при разрешении 2500, а точное значение массы даст ответ - какой из газов регистрируется. Измерение точной массы доступно на приборах с двойной фокусировкой, на тандемном квадрупольном масс-спектрометре TSQ Quantum и на масс-спектрометрах ионно-циклотронного резонанса.
Динамический диапазон. Если мы анализируем смесь, содержащую 99.99 % одного соединения или какого-либо элемента и 0.01% какой-либо примеси, мы должны быть уверены, что правильно определяем и то и другое. Для того, чтобы быть уверенным в определении компонентов в этом примере, нужно иметь диапазон линейности в 4 порядка. Современные масс-спектрометры для органического анализа характеризуются динамическим диапазоном в 5-6 порядков, а масс-спектрометры для элементного анализа 9-12 порядков. Динамический диапазон в 10 порядков означает, что примесь в пробе будет видна даже тогда, когда она составляет 10 миллиграмм на 10 тонн.
Чувствительность. Это одна из важнейших характеристик масс-спектрометров. Чувствительность это величина, показывающая какое количество вещества нужно ввести в масс-спектрометр для того, чтобы его можно было детектировать. Для простоты будем рассматривать связанный с чувствительностью параметр - минимальное определяемое количество вещества, или порог обнаружения. Типичная величина порога обнаружения хорошего хромато-масс-спектрометра, используемого для анализа органических соединений, составляет 1 пикограмм при вводе 1 микролитра жидкости. Давайте представим себе что это такое. Если мы наберем специальным шприцом 1 микролитр жидкости (одна миллионная доля литра) и выпустим ее на листок чистой белой бумаги, то при ее рассмотрении в лупу мы увидим пятнышко, равное по размерам следу от укола тонкой иглой. Теперь представим себе, что мы бросили 1 грамм вещества (например, одну таблетку аспирина) в 1000 тонн воды (например, бассейн длиной 50 метров, шириной 10 метров и глубиной 2 метра). Тщательно перемешаем воду в бассейне, наберем шприцом 1 микролитр этой воды и заколем в хромато-масс-спектрометр. В результате анализа мы получим масс-спектр, который мы сможем сравнить с библиотечным спектром и методом отпечатков пальцев убедиться в том, что это действительно ацетилсалициловая кислота, иначе называемая аспирином.
Пределы обнаружения неорганических веществ, например, методом ICP/MS (FINNIGAN ELEMENT2) еще более впечатляющие. Здесь бассейн уже будет маловат для приготовления раствора с концентрацией, соответствующей пределу обнаружения. Предел обнаружения для FINNIGAN ELEMENT2 по ряду металлов составляет 1 ppq (одна доля на квадриллион). Это значит, что чувствительности прибора достаточна, чтобы детектировать 1 килограмм металла (например, ртути, свинца и т.д.) растворенного в озере Байкал (при условии его перемешивания и полного растворения)!
В масс-спектрометрии изотопов, например, достаточно 800 - 1000 молекул диоксида углерода (СО2, углекислый газ) чтобы получить сигнал углерода. Для того, чтобы продемонстрировать, с какими точностями и изотопными чувствительностями имеет дело изотопная масс-спектрометрия, прибегнем к следующей аллегории. Предположим на одну тысячу совершенно одинаковых яблок, каждое из которых весит 100 грамм, приходится 11 яблок, весящих на 8 % больше, то есть 108 грамм. Все эти яблоки собраны в одном мешке. Этот пример соотвествует соотношению изотопов углерода в природе - на 1000 атомов 12С приходится 11 атомов 13С. Изотопная масс-спектрометрия измеряет соотношения, то есть она способна различить не просто эти 11 яблок, а найти среди многих мешков те, в которых из 1000 стограммовых яблок не 11 стовосьми граммовых, а 10 или 12. Этот пример очень легок для изотопной масс-спектрометрии, на самом деле такие приборы как FINNIGAN DELTAPlus ADVANTAGE, DELTA Plus XP и FINNIGAN МАТ253способны определить разницу в один изотоп (одно сто восьмиграммовое яблоко) среди десяти миллионов атомов (десяти миллионов яблок).
Важнейшая характеристика при анализе органических соединений - это чувствительность. Для того, чтобы достигнуть как можно большей чувствительности при улучшении отношения сигнала к шуму прибегают к детектированию по отдельным выбранным ионам. Выигрыш в чувствительности и селективности при этом колоссальный, но при использовании приборов низкого разрешения приходится приносить в жертву другой важный параметр - достоверность. Ведь если Вы записывали только один пик из всего характеристического масс-спектра, Вам понадобится еще много поработать, чтобы доказать, что этот пик соответствует именно тому компоненту, который Вас интересует. Как же разрешить эту проблему? Использовать высокое разрешение на приборах с двойной фокусировкой, где можно добиться высокого уровня достоверности не жертвуя чувствительностью. Или использовать тандемную масс-спектрометрию, когда каждый пик, соответствующий одиночному иону можно подтвердить масс-спектром дочерних ионов. Итак, абсолютным рекордсменом по чувствительности является органический хромато-масс-спектрометр высокого разрешения с двойной фокусировкой. Так, например, паспортная характеристика DFS гласит, что 2,3,7,8-тетрахлоро-п-дибензодиоксин, введенный через хроматографическую колонку в количестве 10 фемтограмм даст пик, характеризующийся отношением сигнал/шум = 80: 1. Не достижимый ни на каком другом приборе результат!
По характеристике сочетания чувствительности с достоверностью определения компонентов следом за приборами высокого разрешения идут ионные ловушки. Классические квадрупольные приборы нового поколения (TRACE DSQ II) имеют улучшенные характеристики благодаря ряду инноваций, примененных в них, например, использованию искривленного квадрупольного префильтра, предотвращающего попадание нейтральных частиц на детектор и, следовательно, снижению шума.
Зачем нужна масс-спектрометрия
Глубинные физические законы, передовые научные и инженерные разработки, высокотехнологичные вакуумные системы, высокие электрические напряжения, самые лучшие материалы, высочайшее качество их обработки, современнейшая быстродействующая цифровая и аналоговая электроника и компьютерная техника, изощренное программное обеспечение - вот из чего сложен современный масс-спектрометр. И для чего же все это? Для ответа на один из важнейших вопросов мироздания - из чего сложена материя. Но это вопрос не высокой науки, а каждодневной жизни человека.
Например, разработка новых лекарственных средств для спасения человека от ранее неизлечимых болезней и контроль производства лекарств, генная инженерия и биохимия, протеомика. Масс-спектрометрия дала в руки исследователей инструмент, позволяющий идентифицировать белки, определять какие изменения произошли с их структурой вследствие различных взаимодействий, при их воспроизводстве, определить пути метаболизма различных лекарственных средств и других соединений и идентифицировать метаболиты, разрабатывать новые целевые лекарственные средства. Масс-спектрометрия - единственный метод, решающий все эти и многие другие задачи аналитической биохимии.
Без масс-спектрометрии немыслим контроль над незаконным распространением наркотических и психотропных средств, криминалистический и клинический анализ токсичных препаратов, анализ взрывчатых веществ.
Выяснение источника происхождения очень важно для решения целого ряда вопросов: например, определение происхождения взрывчатых веществ помогает найти террористов, наркотиков - бороться с их распространением и перекрывать пути их трафика. Экономическая безопасность страны более надежна, если таможенные службы могут не только подтверждать анализами в сомнительных случаях страну происхождения товара, но и его соотвествие заявленному виду и качеству. А анализ нефти и нефтепродуктов нужен не только для оптимизации процессов переработки нефти или геологам для поиска новых нефтяных полей, но и для того, чтобы определить виновных в разливах нефтяных пятен в океане или на земле.
В эпоху «химизации сельского хозяйства» весьма важным стал вопрос о присутствии следовых количеств применяемых химических средств (например, пестицидов) в пищевых продуктах. В мизерных количествах эти вещества могут нанести непоправимый вред здоровью человека.
Целый ряд техногенных (то есть не существующих в природе, а появившихся в результате индустриальной деятельности человека) веществ являются супертоксикантами (имеющими отравляющее, канцерогенное или вредное для здоровья человека действие в предельно низких концентрациях). Примером является хорошо известный диоксин.
Существование ядерной энергетики немыслимо без масс-спектрометрии. С ее помощью определяется степень обогащения расщепляющихся материалов и их чистота.
Конечно и медицина не обходится без масс-спектрометрии. Изотопная масс-спектрометрия углеродных атомов применяется для прямой медицинской диагностики инфицированности человека Helicobacter Pylori и является самым надежным из всех методов диагностики.
ВЭЖХ/МС системы являются основным аналитическим инструментом при разработке новых лекарственных средств. Без этого метода не может обходиться и контроль качества производимых лекарств и выявления такого распространенного явления как их фальсификация.
Протеомика дала в руки медицины возможность сверхранней диагностики самых страшных заболеваний человечества - раковых опухолей и кардиологических дисфункций. Определение специфических белков, называемых биомаркерами, позволяет проводить раннюю диагностику в онкологии и кардиологии.
Трудно представить область человеческой деятельности, где не нашлось бы места масс-спектрометрии. Ограничимся просто перечислением: биохимия, клиническая химия, общая химия и органическая химия, фармацевтика, косметика, парфюмерия, пищевая промышленность, химический синтез, нефтехимия и нефтепереработка, контроль окружающей среды, производство полимеров и пластиков, медицина и токсикология, криминалистика, допинговый контроль, контроль наркотических средств, контроль алкогольных напитков, геохимия, геология, гидрология, петрография, минералогия, геохронология, археология, ядерная промышленность и энергетика, полупроводниковая промышленность, металлургия.
Данный метод принципиально отличается от рассмотренных выше спектроскопических методов. Структурная масс-спектрометрия основана на разрушении органической молекулы в результате ионизации тем или иным способом.
Образующиеся ионы сортируются по величинам их отношения масса/заряд (m/z), затем регистрируется число ионов для каждого значения этого отношения в виде спектра. На рис. 5.1. представлена общая схема типичного масс-спектрометра.

Рис. 5.1. Блок-схема типичного масс-спектрометра
Для ведения пробы в масс-спектрометр обычно применяют какой-либо вид хроматографии, хотя во многих приборах есть возможность для прямого ввода образца в ионизационную камеру. Во всех масс-спектрометрах имеются устройства для ионизации пробы и разделения ионов по величине m/z. После разделения нужно детектировать ионы и измерять их количество. Типичный коллектор ионов состоит из коллимирующих щелей, которые направляют в коллектор в данный момент только ионы одного вида, где они детектируются, а сигнал детектирования усиливается электронным умножителем. Современные масс-спектрометры укомплектованы специализированным программным обеспечением: компьютеры контролируют накопление, хранение и визуализацию данных.
В настоящее время стала обычной практика объединения масс-спектрометра с газовым (ГХ-МС) или жидкостным (ЖХ-МС) хроматографом.
Все масс-спектрометры подразделяются на два класса: приборы низкого (единичного) и высокого разрешения (R). Спектрометры низкого разрешения – приборы, на которых можно разделить целые массы до m/z 3000 (R = 3000/(3000-2990) = 3000). На таком приборе соединения C 16 H 26 O 2 и С 15 Н 24 NO 2 неразличимы, поскольку прибор будет фиксировать и в первом и во втором случае массу 250.
Приборы высокого разрешения (R = 20000) смогут различить соединения C 16 H 26 O 2 (250.1933) и С 15 Н 24 NO 2 (250.1807), в этом случае R = 250.1933/(250.1933 – 250.1807) = 19857.
Таким образом, на приборах низкого разрешения можно устанавливать структурную формулу вещества, однако зачастую для этой цели дополнительно необходимо привлекать данные других методов анализа (ИК-, ЯМР-спектроскопия).
Приборы высокого разрешения могут измерять массу иона с точностью, достаточной для определения атомного состава, т.е. определять молекулярную формулу исследуемого вещества.
В последнее десятилетие происходило быстрое развитие и совершенствование масс-спектрометров. Не обсуждая их устройство, отметим, что они подразделяются по типам в зависимости от 1) способа ионизации, 2) метода разделения ионов. В общем, способ ионизации не зависит от метода разделения ионов и наоборот, хотя имеются исключения. Более полная информация по данным вопросам изложена в литературе [Сайнсб. Лебедев].
В данном пособии будут рассмотрены масс-спектры, полученные ионизацией электронным ударом.
5.2. Масс-спектры с ионизацией электронным ударом
Электронный удар (ЭУ, electron impact, EI) – наиболее распространенный метод ионизации в масс-спектрометрии. Преимуществом этого метода является возможность использования поисковых систем и баз данных (метод ЭУ был исторически первым методом ионизации, основные базы экспериментальных данных получены на приборах с ЭУ).
Молекула вещества пробы в газовой фазе подвергается бомбардировке электронов с высокой энергией (обычно 70 эВ) и выбрасывает электрон, образуя катион-радикал, называемый молекулярным ионом :
М + e → М + (молекулярный ион) + 2e
Наименьшая энергия бомбардирующих (ионизующих) электронов, при которой возможно образование из данной молекулы иона, называется энергией (или, менее удачно, «потенциалом») ионизации вещества (U e).
Энергия ионизации является мерой прочности, с какой молекула удерживает наименее сильно связанный с ней электрон.
Как правило, для органических молекул энергия ионизации составляет 9-12 эВ, поэтому бомбардировка электронами с энергией 50 эВ и выше сообщает избыточную внутреннюю энергию возникающему молекулярному иону. Эта энергия частично рассеивается за счет разрыва ковалентных связей.
В результате такого разрыва происходит распад молекулярного иона на частицы меньшей массы (фрагменты). Такой процесс называется фрагментацией .
Фрагментация происходит избирательно, является высоковоспроизводимой и характеристичной для данного соединения . Более того, процессы фрагментации предсказуемы, и именно они обуславливают широкие возможности масс-спектрометрии для структурного анализа. По-сути, структурный анализ методом масс-спектрометрии заключается в идентификации осколочных ионов и ретроспективном восстановлении структуры исходной молекулы, исходя из направлений фрагментации молекулярного иона. Так, например, метанол образует молекулярный ион по схеме:
О дна
точка – оставшийся нечетный электрон;
когда заряд локализован на отдельном
атоме, знак заряда указывается на этом
атоме.
дна
точка – оставшийся нечетный электрон;
когда заряд локализован на отдельном
атоме, знак заряда указывается на этом
атоме.
Многие из этих молекулярных ионов распадаются за время 10 -10 – 10 -3 с и дают ряд осколочных ионов (первичная фрагментация):

Если некоторые из молекулярных ионов имеют достаточно большое время жизни, то они достигают детектора и регистрируются в виде пика молекулярного иона. Поскольку заряд исходного иона равен единице, отношение m / z для этого пика дает молекулярную массу исследуемого вещества.
Таким образом, масс-спектр – это представление относительных концентраций положительно заряженных осколков (включая молекулярный ион) в зависимости от их масс .
В специальной литературе приводятся таблицы наиболее часто встречающихся фрагментных ионов, где указана структурная формула иона и его значение m/z [Преч, Гордон, Сильверстейн].
Высота наиболее интенсивного в спектре пика принимается за 100%, а интенсивности других пиков, включая пик молекулярного иона, выражаются в процентах от максимального пика.
В определенных случаях самым интенсивным может быть и пик молекулярного иона. В общем случае: интенсивность пика зависит от устойчивости образующегося иона .
В масс-спектрах часто присутствует серия пиков фрагментных ионов, различающихся на гомологическую разность (СН 2), т.е. 14 а.е.м. Гомологические серии ионов характерны для каждого класса органических веществ, а потому они несут важную информацию о структуре исследуемого вещества.
Что происходит с образцами крови, которую вы сдаете на клинический анализ? Сколько весит ваш гемоглобин? Каким образом ученые вообще взвешивают молекулы - мельчайшие частицы вещества, которые невозможно увидеть или потрогать? Обо всем этом в рамках рубрики «Просто о сложном» T&P рассказала студентка 5-го курса кафедры химической физики ФМХФ, сотрудница лаборатории ионной и молекулярной физики МФТИ Екатерина Жданова.
Очень часто методы исследований интересуют лишь специалистов в конкретных областях и остаются в тени более фундаментальных проблем, например происхождения жизни или принципов работы человеческого сознания. Тем не менее для поиска ответа на «главный вопрос жизни, Вселенной и всего остального» сначала необходимо научиться отвечать на вопросы более простые. Например, как взвесить молекулу? Обычные весы тут вряд ли помогут: масса молекулы метана - около 10^(-23) грамм. Молекула гемоглобина, крупного и сложного белка, весит в несколько раз больше - 10^(-20) грамм. Ясно, что необходим какой-то иной подход к проблеме, ведь привычные нам измерительные приборы к ней не применимы. Надо также понимать, что, взвешивая в магазине яблоки или становясь на весы после тренировок, мы на самом деле измеряем силу, действующую на прибор - весы. Затем уже происходит пересчет в привычные нам единицы - граммы и килограммы.
Но как же взвесить молекулу? Здесь природа оставила нам лазейку. Оказывается, заряженные частицы «чувствуют» присутствие электрического и магнитного поля и изменяют траекторию и характер своего движения. На заряженные частицы также действуют силы, величину которых можно пересчитать в отношении массы к заряду. Этот метод сегодня довольно популярен и называется масс-спектрометрия. Первооткрывателем масс-спектрометрии считается сэр Дж. Дж. Томсон, нобелевский лауреат по физике. Он обратил внимание на то, что заряженные частицы движутся в магнитном поле по параболическим траекториям, пропорциональным отношению их массы к заряду.
Схема работы масс-спектрометра состоит из нескольких этапов. Прежде всего анализируемое вещество должно пройти ионизацию. Затем оно попадает в систему ионного транспорта, которая должна доставить заряженные частицы в масс-анализатор. В масс-анализаторе как раз происходит разделение ионов в зависимости от отношения массы к заряду. В завершение ионы попадают на детектор, данные с которого анализируются с помощью специального программного обеспечения. Полученная таким образом картинка представляет собой спектр, то есть распределение частиц. Одна из осей этого графика - отношения массы к заряду, вторая - интенсивность. Каждый из пиков на таком графике будет характерным для ионов конкретного вещества, поэтому попадание в прибор посторонних веществ, например воздуха, может привести к искажениям результатов. Чтобы избежать этого, применяется вакуумная система.

Сравнительно простая физическая концепция данного метода требует ряда нетривиальных инженерных решений. Как ионизировать молекулы? Каким способом создавать электромагнитное поле? Атомы и молекулы электрически нейтральны, поэтому для проведения масс-спектрометрических измерений необходимо их ионизировать, то есть оторвать электроны с внешних атомных орбиталей или добавить протон. Важную роль играет тип образца, с которым предстоит работать. Для исследования неорганических веществ - металлов, сплавов, горных пород - необходимо использовать одни методы, для органических веществ подходят другие. Очень многие органические вещества (такие как ДНК или полимеры) сложно испарить, то есть перевести в газ, без разложения, а это значит, что исследования живой ткани или биологических образцов требуют применения специальных методов. Кроме того, при ионизации молекулы могут распадаться на отдельные фрагменты. Так мы снова встаем перед вопросом: что именно мы собираемся измерить? Массу всей молекулы или массу фрагментов? И то и другое важно. Более того, измерив массу целой молекулы, исследователи часто специально дробят ее на куски. Так, определив массу структурных элементов белка, мы вместе с тем определяем и их количество, что позволяет нам делать выводы о его химическом составе и структуре.
Все это говорит о разнообразии существующих масс-спектрометров, каждый из которых применяется для решения задач в конкретной области. Этот метод практически незаменим в тех случаях, когда ученым необходимо определить химический состав вещества. Фармацевты применяют масс-спектрометрические эксперименты при разработке лекарств, исследованиях фармакокинетики (то есть биохимических процессов, происходящих в организме при принятии лекарства) и метаболизма. Ученые-биологи используют масс-спектрометрию для анализа белков, пептидов и нуклеиновых кислот. Кроме того, если мы хотим проверить качество воды или продуктов питания, то нам снова не обойтись без этого метода.
Отдельная инновационная область применения масс-спектрометрии - медицинская диагностика. К развитию множества заболеваний приводят структурные изменения белков нашего организма: обычно они классифицируются по образованию характерного кусочка, пептида-маркера. Если вовремя определить такую мутацию, то появляется возможность лечить болезнь на ранней стадии. Кроме того, благодаря современным масс-спектрометрам становится возможным проводить исследования такого рода в режиме реального времени - например, в ходе нейрохирургической операции. Это позволяет точно определять границы между здоровой тканью и опухолью, что критически важно для хирургов.
Кажущаяся на первый взгляд сухой и узкопрофильной, масс-спектрометрия при внимательном ознакомлении оказывается удивительно богатой областью, объединяющей широкий класс приложений с необычными инженерными решениями. Наука показывает, что ответы на менее фундаментальные вопросы порой не менее интересны.
ЧЕЛЯБИНСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ
Химический факультет
Курсовая работа на тему
«Масс-спектрометрический метод анализа»
Выполнил: студент группы Х-202
Меньшенин А.Н.
Проверила: Данилина Е.И.
Челябинск
2007
Содержание
ВВЕДЕНИЕ
Основы масс-спектрометрии
Принципиальное устройство масс-спектрометра
Способы ввода образца
Механизмы ионизации
Протонирование
Депротонирование
Катионизация
Отрыв электрона
Захват электрона
Способы ионизации
Ионизация электроспрея (ESI)
1. Растворители для электроспрея
2. Устройство прибора ионизации электроспрея
Ионизация наноэлектроспрея (nanoESI)
Химическая ионизация при атмосферном давлении (APCI)
Фотоионизация при атмосферном давлении (APPI)
Лазерная десорбция/ионизация при помощи матрицы (MALDI)
3. Преимущества и недостатки метода лазерной десорбции/ионизации при помощи матрицы (MALDI).
Десорбция/ионизация на кремнии (DIOS)
Бомбардировка быстрыми атомами/ионами (FAB)
Электронная ионизация (EI)
Химическая ионизация (CI)
Сравнение основных характеристик способов ионизации
Анализаторы масс
Анализ масс
Краткий обзор принципов работы анализаторов
Рабочие характеристики анализаторов
4. Точность
5. Разрешение (разрешающая сила)
6. Диапазон масс
7. Тандемный анализ масс (MS/MS или MS n)
8. Скорость сканирования
Конкретные виды анализаторов
Квадрупольный анализатор
Квадрупольная ионная ловушка
Линейная ионная ловушка
9. Ограничения ионной ловушки
Двуфокусирующий магнитный сектор
Квадрупольная-времяпролётная тандемная масс-спектрометрия
10. MALDI и времяпролётный анализ
Квадрупольная времяпролётная масс-спектрометрия
Масс-спектрометрия с Фурье-преобразованием (FTMS)
Общее сравнение анализаторов масс, обычно используемых совместно с ES
Детекторы
Электронный умножитель
Цилиндр Фарадея
Фотоумножитель с преобразующим динодом
Матричный детектор
Зарядовый (индуктивный) детектор
Общее сравнение детекторов.
Вакуум масс-спектрометра
Список использованной литературы
ВВЕДЕНИЕ
Масс-спектрометрию описывали как мельчайшие весы в мире, не из-за размера масс-спектрометра, но из-за того, что он взвешивает – молекулы. За последнее время масс-спектрометрия претерпела потрясающий технологический подъём, позволяющий применять её для белков, пептидов, углеводов, ДНК, лекарств и многих других биологически активных молекул. Благодаря таким способам ионизации, как ионизация электроспрея (ESI) или лазерная десорбция/ионизация из матрицы (MALDI), масс-спектрометрия стала незаменимым инструментом для биохимических исследований.
Основы масс-спектрометрии
Масс-спектрометр определяет массу молекулы, измеряя отношение массы к заряду (m / z ) её иона. Ионы генерируются при потере или получении заряда нейтральными частицами. После образования ионы электростатически направляются в анализатор массы, где они разделяются соответственно своему m / z и, наконец, детектируются. Результатом ионизации молекул, разделения ионов и детектирования ионов является спектр, по которому можно определить молекулярную массу и даже некоторую информацию о строении вещества. Можно провести аналогию между масс-спектрометром и призмой, как показано на рис. 1.1 . В призме свет разделяется на компоненты по длинам волн, которые затем определяются оптическим рецептором. Точно так же, в масс-спектрометре сгенерированные ионы разделяются в анализаторе массы, подсчитываются и определяются в детекторе ионов (таких, как, например, электронный умножитель).
ции, анализатор массы и детектор ионов. Некоторые приборы комбинируют ввод образца и ионизацию, в других объединены анализатор массы и детектор. Однако все молекулы образца претерпевают одинаковые воздействия независимо от конфигурации прибора. Молекулы образца вводятся через систему впуска. Попав внутрь прибора, молекулы преобразуются в ионы в устройстве ионизации, а затем электростатически переносятся в анализатор массы. Ионы затем разделяются соответственно их m / z . Детектор преобразует энергию ионов в электрические сигналы, которые затем поступают в компьютер.
Способы ввода образца
Ввод образца был одной из первых проблем в масс-спектрометрии. Для проведения анализа масс образца, который первоначально находится при атмосферном давлении (760 Торр), он должен быть введён в прибор таким образом, чтобы вакуум внутри последнего остался практически неизменным (~10 -6 Торр). Основными методами ввода образца являются прямое введение
зонда или подложки, обычно используемое в MALDI-MS, или прямое вливание или впрыскивание в устройство ионизации, как в методе ESI-MS.

Прямое введение : использование прямого введения зонда/подложки (рис. 1.3 ) – очень простой способ доставки образца в прибор. Образец сначала размещается на зонде, а затем вводится в ионизационную зону масс-спектрометра, обычно через вакуумный клапан. Образец после подвергается необходимым процедурам десорбции, таким как лазерная десорбция или прямое нагревание, чтобы обеспечить испарение и ионизацию.
Прямое вливание или впрыскивание : простой капилляр или капиллярная колонка используется для помещения образца в газообразной форме или в растворе. Прямое вливание также удобно, потому что оно позволяет эффективно вводить малые количества вещества в масс-спектрометр без нарушения вакуума. Капиллярные колонки обычно используются для разграничения систем разделения и устройства ионизации масс-спектрометра. Эти системы, включая газовую хроматографию (ГХ) и жидкостную хроматографию (ЖХ), также служат для разделения различных компонентов раствора, важных для анализа масс. В газовой хроматографии разделение различных компонентов происходит в стеклянной капиллярной колонке. Как только пары образца покидают хроматограф, они направляются прямиком в масс-спектрометр.

В 1980-х годах невозможность совместного использования жидкостной хроматографии (ЖХ) с масс-спектрометрией была обусловлена, большей частью, неспособностью устройств ионизации справляться с непрерывным по-
током ЖХ. Однако, ионизация электроспрея (ESI), химическая ионизация при атмосферном давлении (APCI) и фотоионизация при атмосферном давлении (APPI) сейчас позволяют совмещать ЖХ и масс-спектрометрию в повседневных анализах.
Механизмы ионизации
Протонирование – механизм ионизации, при котором к молекуле присоединяется протон, сообщая ей заряд 1+ на каждый присоединённый протон. Положительные заряды обычно локализуются на основных частях молекулы, таких, как амины, с образованием стабильных катионов. Пептиды часто ионизируются при помощи протонирования. Протонирование осуществляется при MALDI, ESI и APCI.
Депротонирование– механизм ионизации, при котором отрицательный заряд 1- получается при отрыве протона от молекулы. Такой механизм ионизации обычно осуществляется при MALDI, ESI и APCI и очень полезен для определения кислотных образцов, включая фенолы, карбоновые кислоты и сульфоновые кислоты. Спектр отрицательных ионов сиаловой кислоты показан на рис 1.2 .
Катионизация – механизм ионизации, в котором заряженный комплекс образуется при координационном присоединении положительно заряженного иона к нейтральной молекуле. В принципе, пртонирование тоже подпадает под это определение, поэтому катионизацией считается присоединение иона, отличного от протона, например щелочного металла или аммония. Кроме того, катионизация применима к молекулам, которые неспособны к протонированию. Связь катионов, в отличие от протонов, с молекулой менее ковалентна, поэтому заряд остаётся локализован на катионе. Это минимизирует размывание заряда и фрагментацию молекулы. Катионизация также может быть произведена при MALDI, ESI и APCI. Углеводы – лучшие вещества для такого механизма ионизации, с Na + как обычным присоединённым катионом.
Прямой перенос заряженной молекулы в газовую фазу
Перенос соединений, уже заряженных в растворе, легко достигается при использовании десорбции или выбрасыванием заряженных частиц из конденсированной фазы в газовую. Обычно это осуществляется с использованием MALDI или ESI.
Отрыв электрона
Как видно из названия механизма, отрыв электрона придаёт молекуле 1+ положительный заряд при выбивании электрона, так что при этом часто образуются катион-радикалы. Наблюдаемый, в основном, при электронной ионизации, отрыв электрона обычно применяется для относительно неполярных соединений с низкой молекулярной массой. Также известно, что он часто приводит к образованию значительных количеств фрагментарных ионов.
Захват электрона
При захвате электрона, отрицательный заряд 1- сообщается молекуле при присоединении электрона. Этот механизм ионизации в первую очередь наблюдается для молекул с большим сродством к электрону, таких как галогенсодержащие соединения.
Масс-спектрометрия - это метод измерения отношения массы заряженных частиц к их заряду (m/z).
Для проведения масс-спектрометрического анализа образец переводят в ионизированную форму. После этого тем или иным способом производится разделение ионов по отношению их массы к зарядам и регистрация этих ионов, которые могут быть как положительными, так и отрицательными.
Масс-спектрометрический анализ дает важную информацию для определения молекулярной массы, молекулярной формулы или элементного состава и структуры молекул.
Масс-спектрометрию используют для определения относительной молекулярной массы М г соединения, которую выражают в атомных единицах массы (а.е.м.) или дальтонах, Да, (1 Да = 1 а.е.м.=1,660541 - 10 -27 кг, что равно 1/12 массы изотопа углерода с массовым числом 12). Масса основного изотопа углерода 12 С выражается целым числом и равна 12,000000 Да. Массы всех изотопов любых других элементов будут выражаться нецелыми числами.
В масс-спектре пики или линии с определенным отношением m/z, соответствуют молекулярным фрагментам и также обозначаются целым числом, полученным при округлении точного значения m/z.
В масс-спектрометрии существует три различных понятия массы. Средняя молекулярная масса вычисляется на основании элементного состава и средних атомных масс. Средняя молекулярная масса важна при изучении больших молекул. Номинальная молекулярная масса вычисляется с учетом элементного состава и номинальных атомных масс наиболее распространенных в природе изотопов. Точная молекулярная масса вычисляется из значений точных масс наиболее распространенных изотопов.
С помощью масс-спектрометрии возможны: анализ органических соединений, неорганический анализ, исследования по выяснению механизмов реакций в органической химии и анализ поверхности.
С помощью масс-спектрометрии как аналитического метода решают громадное число качественных и количественных задач. Качественные исследования заключаются в определении структуры неизвестного соединения, в частности, природных веществ, метаболитов лекарственных препаратов и других ксенобиотиков, синтетических соединений. Для количественного анализа масс-спектрометрию используют при разработке арбитражных методов и методов сравнения. Масс-спектрометрия сегодня развивается очень быстро, охватывая все более широкие области применения. Объединение масс-спектрометрии с хроматографией значительно увеличило возможности метода и расширило круг изучаемых объектов.
5.12. Электрогравиметрия
В электрогравиметрическом анализе определяемое вещество количественно выделяют из раствора электролизом и по массе выделившегося металла или его оксида на электроде рассчитывают содержание определяемого элемента в пробе.
Электролизом называют химическое разложение вещества под действием электрического тока. На катоде происходит восстановление:
Cu 2+ + 2e → Cu 0
а на аноде – окисление:
2Cl - - 2e → Cl 2 (г) и 2OH - - 2e → 1\2O 2 + H 2 O
Под действием приложенного напряжения заряженные частицы (ионы) перемещаются к электродам. Однако их разряд, т. е. электролиз, начинается при достижении определенной величины напряжения, называемой напряжением разложения
где E а, E к – ЭДС гальванического элемента;
iR – омическое падение напряжения;
η – перенапряжение анода и катода при выделении продуктов электролиза.
Схема установки для проведения электролиза приведена на рис. 5.14.
Электролиз чаще всего проводят при постоянном токе. Для получения постоянного тока обычно используют выпрямитель переменного тока или батарею аккумуляторов 1. Скользящий контакт 2 позволяет регулировать подаваемое напряжение, которое измеряют вольтметром V. Сила тока контролируется амперметром А. При выделении металлов катод 5 обычно используют в виде платиновой сетки, анод 4 – в виде платиновой спирали или пластинки. При выделении оксидов знаки электродов меняются: платиновая сетка становится анодом, а спираль – катодом. Раствор перемешивается механической или магнитной мешалкой 3.

Рис. 5.14. Схема установки для проведения электролиза: 1 – источник постоянного тока; 2 – переменное сопротивление (реостат); 3 – магнитная мешалка;
4 – анод; 5 – катод
В электрогравиметрических методах анализа кроме потенциала, силы тока важно контролировать ряд экспериментальных условий.
5.13. Кулонометрия
В кулонометрических методах определяют количество электричества, которое расходуется в ходе электрохимической реакции. Различают прямую кулонометрию и кулонометрическое титрование.
В методах прямой кулонометрии анализируемое вещество непосредственно подвергается электрохимическому превращению в кулонометрической ячейке (процесс проводят при постоянном контролируемом потенциале) (Рис. 5.15.).

Рис. 5.15. Схема установки для прямой кулономeтрии при постоянном E:
1 - электролизер; 2 - источник постоянного токa с регулируемым напряжением: 3 - прибор для определения количества злектричества: 4 - рабочий электрод; 5 - вспомогательный электрод; 6 - электрод сравнения, относительно которого контролируют потенциал рабочего электрода: 7 - устройство, измеряющее разность потенциалов.
В методе кулонометрического титрования определяемое вещество реагирует с титрантом, который производится в кулонометрической ячейке посредством электролиза специально подобранного раствора.
Кулонометрическое титрование проводят при постоянном токе.
Кулонометрические методы основаны на законах Фарадея. Необходимым условием количественного определения является 100%-й выход по току. Выход по току определяется отношением количества вещества, выделившегося в процессе электролиза, к теоретическому количеству, вычисленному на основании закона Фарадея. Не 100%-й выход по току может быть обусловлен затратами тока на побочные процессы:
1) разложение воды на водород и кислород;
2) восстановление или окисление примесей, например, растворенного в воде кислорода;
3) реакция с участием продуктов электролиза;
4) реакция с участием материала электрода (окисление ртути и др).
При проведении кулонометрических определений нужно предусмотреть все условия, обеспечивающие 100%-й выход по току, контроль рН, выбор электродов, разделение катодного и анодного пространства.
5.14. Кондуктометрия
Кондуктометрический метод анализа основан на измерении удельной электропроводности анализируемого раствора.
Электропроводностью называют величину, обратную электрическому сопротивлению R. Единицей измерения электропроводности является сименс (См) или Ом -1 . Растворы электролитов, являясь проводниками II рода, подчиняются закону Ома. По аналогии с сопротивлением проводников I рода сопротивление раствора прямо пропорционально расстоянию между электродами d и обратно пропорционально площади их поверхности A:
где р - удельное сопротивление, Ом · см.
При d =1 см иА =1 см 2 имеем R = р, следовательно, удельное сопротивление равно сопротивлению 1 см 3 раствора.
Величину, обратную удельному сопротивлению, называют удельной электропроводностью:
Удельная электропроводность (См ∙ см -1) численно равна току (А), проходящему через слой раствора с поперечным сечением, равным единице, под действием градиента потенциала 1 В на единицу длины.
Электропроводность разбавленных растворов электролитов зависит от числа ионов в растворе (т. е. от концентрации), числа элементарных зарядов, переносимых каждым ионом (т. е. от заряда иона), и от скорости движения одинаково заряженных ионов к катоду или аноду под действием электрического поля (Рис. 5.16.). С учетом всех этих факторов электропроводящие свойства ионов характеризуют эквивалентной ионной электропроводностью (подвижностью).

Рис. 5.16. Кондуктометр ОК 102/1: 1 – корпус прибора; 2 – измерительная
шкала; 3 – тумблер «Сеть»; 4 - переключатель пределов измерения «Range»; 5 – ручка калибровки потенциометра «Calibration»; 6 – кнопка калибровки «Calibration».
Различают прямую и косвенную кондуктометрию, или кондуктометр ическое титрование.
Прямая кондуктометрия мало применяется в аналитической химии. Причина этого в том, что электропроводность является величиной аддитивной и определяется присутствием всех ионов в растворе. Прямые кондуктометрические измерения используют для контроля качества воды, применяемой в химической лаборатории, и современные установки для перегонки или деминерализации воды снабжаются кондуктометрическими датчиками - кондуктометрами для измерения удельной электропроводности растворов. Детекторы по электропроводности применяются в ионной хроматографии.
К достоинствам метода кондуктометрического титрования относится возможность высокоточных измерений даже в очень разбавленных растворах.
Для кондуктометрического титрования пригодны кислотно-основные или осадительные реакции, сопровождающиеся заметным изменением электропроводности вследствие образования малодиссоциирующих или малорастворимых соединений.
5.15. Титриметрия
Титриметрический анализ (титрование) - метод количественного/массового анализа, который часто используется в аналитической химии, основанный на измерении объёма раствора реактива точно известной концентрации, расходуемого для реакции с определяемым веществом (Рис. 5.17.).

Рис. 5.17. Настольный электрохимический прибор
OHAUS Starter 2100
Титрование - процесс определения титра исследуемого вещества. Титрование производят с помощью бюретки, заполненной титрантом до нулевой отметки. Титровать начиная от других отметок не рекомендуется, так как шкала бюретки может быть неравномерной. Заполнение бюреток рабочим раствором производят через воронку или с помощью специальных приспособлений, если бюретка полуавтоматическая. Конечную точку титрования (не следует путать с точкой эквивалентности) определяют индикаторами или физико-химическими методами (по электропроводности, светопропусканию, потенциалу индикаторного электрода и т. д.). По количеству пошедшего на титрование рабочего раствора рассчитывают результаты анализа.
Методы титрования
Процесс титрования сопровождается изменением равновесных концентраций реагента, определяемого вещества и продуктов реакции. Это удобно изобразить графически в виде т. наз. кривой титрования в координатах концентрация определяемого.вещества (или пропорциональная ей величина)-объем (масса) титранта.
(1) Косвенное титрование, или титрование заместителя – титрование, которое применяют, когда нет подходящей реакции или индикатора для прямого титрования. В этом случае используют реакцию, в которой анализируемое вещество замещают эквивалентным количеством другого вещества и затем титруют рабочим раствором.
(2) Метод объемного (титрометрического) анализа - это метод количественного определения, основанный на измерении объема реагента, требуемого для проведения реакции с определяемым веществом.
(3) Обратное титрование – титрование, которое используют в тех случаях, когда прямое титрование невозможно или когда анализируемое вещество неустойчиво. При этом берут два рабочих раствора, один из которых добавляют в избытке, а вторым титруют избыток первого.
(4) Прямое титрование наиболее распространенный и удобный прием, когда к анализируемому раствору вещества непосредственно добавляют рабочий раствор известной концентрации.
(5) Титрование –процесс постепенного добавления раствора точно известной концентрации к исследуемому раствору.
(6) Точка эквивалентности – установление конечной точки титрования.
Объемные методы анализа . Титрование как метод количественного определения вещества: прямое, косвенное и обратное
Метод объемного (титрометрического) анализа (2) это метод количественного определения, основанный на измерении объема реагента, требуемого для проведения реакции с определяемым веществом.
Объемные методы анализа основаны на протекании реакций нейтрализации, осаждения, ионного обмена, комплексообразования, окисления-восстановления и др. Они должны удовлетворять следующим условиям:
Строгое соблюдение стехиометрических соотношений между веществами реакций;
Быстрое и количественное протекание реакций;
Точное и строгое фиксирование точки эквивалентности;
Посторонние вещества в анализируемой пробе не должны вступать в реакцию с добавляемым реагентом, что может помешать титрованию.
Титрованием (5)называют процесс постепенного добавления раствора точно известной концентрации к исследуемому раствору.
Одной из основных стадий этого процесса, во многом определяющей точность объемного метода, является установление конечной точки титрования, называемой точкой эквивалентности (6). Точку эквивалентности определяют визуально по изменению цвета раствора, индикатора, появлению помутнения либо инструментальными методами кондуктометрическое, потенциометрическое титрование.
Для титрования достаточно 1-3 капель раствора индикатора массовой долей 0,1-0,5 % на 10-100 см 3 анализируемого раствора.
Титрометрическое определение осуществляют прямым, косвенным и обратным титрованием.
Прямое титрование (4)наиболее распространенный и удобный прием, когда к анализируемому раствору вещества непосредственно добавляют рабочий раствор известной концентрации.
Косвенное титрование, или титрование заместителя (1), применяют, когда нет подходящей реакции или индикатора для прямого титрования. В этом случае используют реакцию, в которой анализируемое вещество замещают эквивалентным количеством другого вещества и затем титруют рабочим раствором.
Обратное титрование (3) используют в тех случаях, когда прямое титрование невозможно или когда анализируемое вещество неустойчиво. При этом берут два рабочих раствора, один из которых добавляют в избытке, а вторым титруют избыток первого.
Расчет массовой доли определяемого вещества Х (в %) через массовую концентрацию рабочего раствора ведут по формуле
Х=100 VСМ /(1000т), (5.5)
где V - объем рабочего раствора, пошедшего на титрование, см 3 ;
С -молярная концентрация рабочего раствора, моль/дм 3 ;
М - молекулярная эквивалентная масса определяемого вещества, г/моль;
m - масса навески анализируемого вещества, г.
6. ВИДЫ ДЕФЕКТОВ МЕТАЛЛА
6.1. Классификация дефектов
Дефектом называют каждое отдельное несоответствие продукции требованиям, установленным нормативной документацией (ГОСТ, ОСТ, ТУ и т.д.). К несоответствиям относятся нарушение сплошности материалов и деталей, неоднородность состава материала: наличие включений, изменение химического состава, наличие других фаз материала, отличных от основной фазы, и др.
Дефектами являются также любые отклонения параметров материалов, деталей и изделий от заданных, таких, как размеры, качество обработки поверхности, влаго- и теплостойкость и ряд других физических величин.
Дефекты подразделяются на явные (те, что выявляются глазами) и скрытые (внутренние, подповерхностные, неразличимые глазом).
В зависимости от возможного влияния дефекта на служебные свойства детали дефекты могут быть:
Критическими (дефекты, при наличии которых использование продукции по назначению невозможно или исключается по соображениям безопасности и надёжности);
Значительными (дефекты, существенно влияющие на использование продукции и/или на её долговечность, но не являющиеся критическими);
Малозначительными (не оказывают влияния на работоспособность продукции).
По происхождению дефекты изделий подразделяют на производственно-технологические (металлургические, возникающие при отливке и прокатке, технологические, возникающие при изготовлении, сварке, резке, пайке, клепке, склеивании, механической, термической или химической обработке и т.п.); эксплуатационные (возникающие после некоторой наработки изделия в результате усталости материала, коррозии металла, изнашивания трущихся частей, а также неправильной эксплуатации и технического обслуживания) и конструктивные дефекты, являющиеся следствием несовершенства конструкции из-за ошибок конструктора.
C целью выбора оптимальных методов и параметров контроля производится классификация дефектов по различным признакам: по размерам дефектов, по их количеству и форме, по месту расположения дефектов в контролируемом объекте и т.д.
Размеры дефектов a могут изменяться от долей миллиметров до сколь угодно большой величины. Практически размеры дефектов лежат в пределах 0,01 мм ≤ a ≤ 1 см.
В ультразвуковой дефектоскопии, например, величина а влияет на выбор рабочей частоты.
При количественной классификации дефектов различают три случая (рис. 6.1): а – одиночные дефекты, б – групповые (множественные) дефекты, в – сплошные дефекты (обычно в виде газовых пузырей и шлаковых включений в металлах).

Рис. 6.1. Количественная классификация дефектов: а – одиночные;
б – групповые; в – сплошные
При классификации дефектов по форме различают три основных случая (рис. 6.2): а – дефекты правильной формы, овальные, близкие к цилиндрической или сферической форме, без острых краёв; б – дефекты чечевицеобразной формы, с острыми краями; в – дефекты произвольной, неопределённой формы, с острыми краями – трещины, разрывы, посторонние включения.
Форма дефекта определяет его опасность с точки зрения разрушения конструкции. Дефекты правильной формы, без острых краёв, наименее опасны, т.к. вокруг них не происходит концентрации напряжений. Дефекты с острыми краями, как на рис. 6.2, б и в, являются концентраторами напряжений. Эти дефекты увеличиваются в процессе эксплуатации изделия по линиям концентрации механических напряжений, что, в свою очередь, приводит к разрушению изделия.

Рис. 6.2. Классификация дефектов по форме: а – правильная форма;
б – чечевицеобразная форма с острыми краями; в – произвольная,
неопределённая форма с острыми краями
При классификации дефектов по положению различают четыре случая (рис. 6.3): а – поверхностные дефекты, расположенные на поверхности материала, полуфабриката или изделия, – это трещины, вмятины, посторонние включения; б – подповерхностные дефекты – это дефекты, расположенные под поверхностью контролируемого изделия, но вблизи самой поверхности; в – объёмные дефекты – это дефекты, расположенные внутри изделия.
Наличие фосфовидных и нитридных включений и прослоек может привести к образованию дефектов четвертого вида – сквозных.
По форме поперечного сечения сквозные дефекты бывают круглые (поры, свищи, шлаковые включения) и щелевидные (трещины, непровары, дефекты структуры, несплошности в местах расположения оксидных и других включений и прослоек).
По величине эффективного диаметра (для дефектов округлого сечения) или ширине раскрытия (для щелей, трещин) сквозные дефекты подразделяются на обыкновенные (> 0,5 мм), макрокапиллярные (0,5…2·10 -4 мм) и микрокапиллярные (< 2·10-4 мм).

Рис. 6.3. Классификация дефектов по положению в контролируемом
объекте: а – поверхностные; б – подповерхностные; в – объёмные
По характеру внутренней поверхности сквозные дефекты подразделяются на гладкие и шероховатые. Относительно гладкой является внутренняя поверхность шлаковых каналов. Внутренняя поверхность трещин, непроваров и вторичных поровых каналов, как правило, шероховатая.
Положение дефекта влияет как на выбор метода контроля, так и на его параметры. Например, при ультразвуковом контроле положение дефекта влияет на выбор типа волн: поверхностные дефекты лучше всего определяются рэлеевскими волнами, подповерхностные – головными волнами, а объёмные – объёмными (продольными) волнами.
Опасность влияния дефектов на работоспособность зависит от их вида, типа и количества. Классификация возможных дефектов в изделии позволяет правильно выбрать метод и средства контроля.
6.2. Производственно-технические дефекты
Дефекты в металлах образуются главным образом при плавлении, при обработке металла давлением (ковка, штамповка и прокат) и при шлифовании.
По ГОСТ 19200-80 дефекты отливок из чугуна и стали подразделяют на пять основных групп. Необходимо отметить, что принятая терминология широко используется также для отливок из сплавов на основе алюминия, магния, титана и других и поэтому может рассматриваться как универсальная.
6.2.1. Литейные дефекты
Несоответствие по геометрии.
Эта группа объединяет 14 видов дефектов, обусловленных нарушением формы, неточностью размеров и массы отливки.
1. Недолил - дефект в виде неполного образования отливки вследствие незаполнения полости формы металлом (рис. 6.4. а). Одной из основных причин недолива является недостаточное количество жидкого металла.
2. Незалив - несоответствие конфигурации отливки чертежу вследствие износа модельной оснастки или дефектов формы (рис. 6.4. б). Причиной незалива может быть также нарушение технологических режимов заливки.
3. Неслитина - сквозная щель или отверстие в стенке отливки, образовавшееся вследствие неслияния встречных потоков металла (рис. 6.4. в). Неслитина характерна для сплавов с широким интервалом кристаллизации и наблюдается обычно в тонких стенках отливок. Эти дефекты легко обнаруживаются при визуальном осмотре отливок.
4. Обжим - это местное нарушение конфигурации отливки вследствие деформации формы при ее сборке или заливке (рис. 6.4. г). Обжим обычно образуется вблизи плоскости разъема в виде прилива или утолщения произвольной формы.
5. Подутость представляет собой местное утолщение отливки, возникшее вследствие расширения недостаточно уплотненной формы заливаемым металлом (рис. 6.4. д).
6-8. Перекос и стержневой перекос - дефекты в виде смещения одной части отливки относительно осей или поверхностей другой части по разъему формы, модели вследствие их неточной установки (рис. 6.4. е) или в виде смещения отверстия, полости или части отливки, выполняемых с помощью стержня, вследствие его перекоса (рис. 6.4. ж). Эти дефекты вызваны неточной фиксацией опок или перекосом стержня при его установке. В последнем случае возникает также разностенность - увеличение или уменьшение толщины стенок отливки (рис. 6.4. з). Разностенность выявляется визуально или с помощью измерительных средств.
9. Стержневой залив - дефект в виде залитого металлом отверстия или полости в отливке, возникающий из-за непроставленного в литейной форме стержня или его обрушения (рис. 6.4. и).
10. Коробление - искажение конфигурации отливки под влиянием напряжений, возникающих при охлаждении отливки или вследствие деформации модельной оснастки. Коробление может проявляться в различных формах, наиболее характерным является появление вогнутости или выпуклости на плоских поверхностях отливок (рис. 6.4. к). Дефект выявляется с помощью измерительных средств. Стрела прогиба 6 может служить мерой коробления.
11. Вылом и зарез - дефекты в виде нарушений конфигурации отливки при выбивке стержней, обрубке литников (рис. 6.4. л), зачистке отливок или их транспортировании.
12. Прорыв и уход металла - дефекты, вызванные вытеканием металл.* из формы вследствие ее недостаточной прочности или слабого крепления ее частей. При этом либо происходит неполное заполнение полости формы с одновременным образованием приливов произвольной формы, либо возникает дефект в виде пустоты в теле отливки, ограниченной тонкой коркой затвердевшего металла (рис. 6.4. м).

Рис. 6.4. Дефекты отливок - несоответствие по геометрии (стрелки указывают на расположение дефекта)